Генная гемофилия что это
Что такое гемофилия
ЧТО ТАКОЕ ГЕМОФИЛИЯ
Гемофилия, шире коагулопатия – заболевание крови, характеризующееся повышенной кровоточивостью, причиной которой является нарушение свертываемости крови.
Нормальная свертываемость крови предотвращает и останавливает кровоизлияния в мышцы и суставы (гемартрозы и гематомы), а также кровотечения при порезах и царапинах, которые могут возникнуть при активной повседневной жизни любого человека.
Свертывание крови – сложный физиологический процесс, в который вовлечено более десятка специальных белков – факторов свертывания крови, обозначаемых римскими цифрами от I до XIII. Дефицит фактора VIII называется гемофилией А, фактора IX – В. Дефицит или дефект (в зависимости от типа и подтипа) фактора Виллебранда называется болезнью Виллебранда. Существуют и более редкие коагулопатии, в частности, дефицит фактора XI – ранее называемый гемофилией С.
История гемофилии
Впервые в письменных источниках о гемофилии упоминается в священной книге иудеев – талмуде. Около 15 веков назад так называемый «вавилонский талмуд» содержит запись об иудейском мальчике, не прошедшем обряд обрезания, так как оба его старших брата и трое двоюродных – по материнской линии умерли от потери крови после такой манипуляции.
В 12 веке Абу аль Касим, врач служивший при дворе одного из арабских правителей Испании, первым описал симптомы гемофилии. Он писал о нескольких семьях, в которых дети мужского пола умирали от небольших повреждений.
Современное научное исследование гемофилии ведется с XIX века. Впервые термин “гемофилия” был введен в 1828 году швейцарским врачом Хопфом.
Первой высокопоставленной носительницей гемофилии считается английская королева Виктория. В “наследство” от нее это заболевание было получено царствующими семьями Германии, Испании и России.
В России этот недуг часто называют “царским”. Он известен из-за болезни Цесаревича Алексея – последнего наследника престола Российской империи.
Проявления гемофилии
Чем тяжелее гемофилия, тем раньше проявляются признаки кровоточивости. Неизбежные падения и ушибы, которые случаются у ребенка, начинающего ходить, могут вызвать синяки на коже и кровотечения из слизистых оболочек губ и языка.
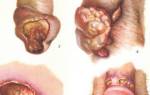
В возрасте 1-3 лет могут начаться поражения мышц и суставов, с болезненными припухлостями, ограничением движений рук и ног. Обширные гематомы, бывающие опасными, могут вызываться внутримышечными инъекциями.
При болезни Виллебранда наиболее частыми симптомами бывают кровотечения из слизистых носа.
Диагностика гемофилии
Иногда наличие диагноза вызывает сомнения, даже при повышенной кровоточивости у самого больного или наличии у других членов семьи гемофилии. Точная диагностика может быть сделана только измерением уровня соответствующего фактора свертывания крови. Такие анализы проводятся в специализированных лабораториях гематологических центров.
Наследование гемофилии
Гемофилия А и В поражает почти исключительно мужчин, а передается по женской линии. По статистике ВОЗ примерно 1 младенец мужского пола из 5000 рождается с гемофилией А, вне зависимости от национальной или расовой принадлежности. Приблизительно одна треть больных гемофилией А не имела у предшествующих поколений подобных расстройств.
Наследование болезни Виллебранда и гипопроконвертинемии не сцеплено с полом.
нажмите, чтобы увеличить
Тяжесть гемофилии
Гемофилию различают по форме тяжести, обусловленной уровнем фактора свертывания крови.
Нормальное содержание FVIII и FIX – 50%-200%.
Гемофилия тяжелой формы. Уровень FVIII или FIX менее 1%. Кровоизлияния в суставы, мышцы и другие органы случаются при минимальных или даже незаметных повреждениях.
Гемофилия средней формы тяжести. Уровень FVIII или FIX 1-5%. Кровотечения возникают из-за явных незначительных повреждений, а также после различных операций и удалений зубов.
Гемофилия легкой формы. Уровень фактора FVIII и FIX 6-30%. Кровоизлияния обычно следуют за крупными повреждениями, хирургическими операциями или удалением зубов. Эта форма может не быть диагностирована достаточно долго.
Тяжесть болезни Виллебранда зависит от её типа и подтипа.
Лечение гемофилии
В прошлом, когда лечение гемофилии производилось устаревшими, не защищенными от вирусов препаратами, инвалидизация больных с тяжелой формой заболевания достигала 90%, а инфицированность гепатитом С – 95%.
Сейчас гемофилию А и В лечат концентратами соответствующих факторов свертывания крови. Комплекты поставки препарата включают в себя лиофилизированный (высушенный) фактор свертывания, воду для растворения, а также приспособления для внутривенных инъекций. Таким образом, имеется возможность для лечения пациента вне медицинского учреждений. Этот тип лечения называется домашним.
Гематологи с раннего детства при тяжелой форме заболевания рекомендуют больным гемофилией профилактическое лечение. При таком лечении, инъекции фактора производятся не по факту кровоизлияния (кровотечения), а вне зависимости от него, по специальному расписанию, составленному врачом-гематологом.
В последние годы в России удалось достичь уровня обеспечения пациентов с гемофилией А и В на уровне развитых стран Запада. Благодаря этому, новое поколение больных гемофилией минимально инвалидизировано. Препараты проходят вирусную инактивацию и шанс заразиться известными на данный момент вирусными инфекциями, передаваемыми через кровь, в современных условиях стремится к нулю.
Генная гемофилия что это
Этиология и встречаемость гемофилии. Гемофилия A (MIM №307600) и гемофилия В (MIM №306900) — Х-сцепленные нарушения свертывания крови, вызываемые мутациями в гене F8 и F9 соответственно. Мутации F8 вызывают недостаточность или дисфункцию фактора свертывания VIII; мутации F9 — фактора IX.
Гемофилия — панэтническое заболевание без расовых различий. Гемофилия А имеет встречаемость 1 на 5 000-10 000 новорожденных мальчиков. Гемофилия В встречается гораздо реже, с частотой 1 на 100 000.
Патогенез гемофилии
Система свертывания поддерживает целостность сосудистой стенки благодаря точному балансу образования и запрещения образования сгустка. Протеазы и белковые кофакторы, формирующие каскад тромбообразования, присутствуют в кровотоке как неактивные предшественники и для того чтобы сформировать сгусток фибрина, должны последовательно активизироваться в месте повреждения.
Своевременное и эффективное образование сгустка требует нарастающей активизации или усиления каскада протеаз. Факторы VIII и IX, соединенные вместе, — ключ к этому усилению; они активизируют фактор свертывания X, а активный фактор X, в свою очередь, еще более активирует факторы IX и VIII. Фактор IX функционирует как протеаза, а фактор VIII — как кофактор. Недостаток или дисфункция фактора IX или VIII вызывает гемофилию.
Мутации в гене F8 представлены делециями, инсерциями, инверсиями и точковыми мутациями. Наиболее частая мутация — инверсия, удаляющая карбоксильный конец фактора VIII; она составляет 25% всех случаев гемофилии А и 40-50% тяжелой гемофилии А. Эта инверсия возникает при внутрихромосомной рекомбинации между последовательностью в 22 интроне гена F8 и гомологичной теломерной последовательности в этом же гене.
Другой интересный класс мутаций — обратная перестановка повторов L1 в гене. Для всех мутаций гена F8 остаточная ферментативная активность комплекса факторов VIII и IX соответствует тяжести клинических проявлений болезни.
У пациентов с гемофилией В описано множество различных мутаций гена F9; но в отличие от распространенной частичной инверсии гена F8 при гемофилии А, частой мутации в гене F9 при гемофилии В не найдено. Необычный вариант F9, фактор Лейдена, вызываемый точковыми мутациями в промоторе гена F9, связан с очень низким содержанием фактора IX и тяжелой гемофилией в детстве, со спонтанным разрешением гемофилии, наступающим в пубертате, когда уровень фактора IX почти нормализуется.
Для всех мутаций в гене F9 остаточная ферментативная активность комплекса факторов VIII—IX также согласуется с тяжестью болезни.
Фенотип и развитие гемофилии
Гемофилия — классическая мужская болезнь, хотя иногда из-за смещения инактивации Х-хромосомы могут поражаться и женщины. Клинически гемофилия А и гемофилия В неразличимы. Оба заболевания характеризуются кровоизлияниями в мягкие ткани, мышцы и крупные суставы. Кровотечение начинается через несколько часов и даже дней после травмы и часто продолжается в течение дней или недель.
Детей с тяжелой формой болезни обычно диагностируют в периоде новорожденности из-за обширных кефалогематом или длительного кровотечения из пупочной ранки. Дети с умеренной формой часто не имеют гематом или гемартрозов, пока не начинают ползать или ходить, и, следовательно, остаются недиагностированными до этого времени. Пациенты с легкой формой болезни часто манифестируют только в юности или взрослом возрасте гемартрозами или длительными кровотечениями после хирургических операций или травм.
Гемофилию А и гемофилию В диагностируют и различают определением активности факторов VIII и IX. Как при гемофилии А, так и В, уровень активности фактора VIII или IX позволяет предсказать клиническую тяжесть болезни.
Особенности фенотипических проявлений гемофилии:
• Возраст начала: от раннего детства до взрослого
• Геморрагический диатез
• Гемартрозы
• Гематомы
Лечение гемофилии
Несмотря на многообещающие исследования по гено-терапии, пока другого, кроме пересадки печени, лечения при гемофилии А и В нет. В настоящее время стандартное лечение — внутривенное введение недостающего фактора. Заместительная терапия повысила ожидаемый срок жизни от 1,4 года в начале прошлого века до почти 65 лет сегодня.
Риски наследования гемофилии
Если женщина имеет гемофилию в семейном анамнезе, можно определить ее статус носительства по анализу сцепления или прямой идентификацией мутаций в гене F8 или F9. Тем не менее обычно идентификация мутаций доступна только для частой инверсии в гене F8. Выявление носительства ферментативным анализом — трудоемкое исследование, и оно широко не доступно.
Если мать — носительница мутации, каждый сын имеет 50% риск гемофилии, а каждая дочь — 50% риск унаследовать мутации F8 или F9. Вследствие низкой частоты клинически значимого смещения инактивации Х-хромосомы, девочки, унаследовавшие мутацию в генах F8 или F9, имеют низкий риск гемофилии.
Если у матери есть сын с гемофилией, но нет других больных родственников, ее априорный риск оказаться носительницей зависит от типа мутации. Точковые мутации и частые инверсии гена F8 почти всегда возникают в мужском мейозе; в результате 98% матерей мальчиков с одной из этих мутаций — носительницы вследствие новой мутации у их отцов (деда больного по матери).
В отличие от этого, мутации типа делеций обычно возникают в женском мейозе. Если тип мутации неизвестен, считают, что приблизительно треть пациентов имеют новую мутацию в F8 или F9. Применяя метод Байеса, этот риск можно модифицировать с учетом числа здоровых сыновей в семье.
Пример гемофилии. Здоровая 38-летняя женщина обратилась на плановую консультацию относительно риска родить ребенка с гемофилией. Ее дядя по матери умер в детстве от гемофилии, у брата в детстве отмечались кровотечения. Проблемы с кровоточивостью у брата закончились в юности. Другие члены семьи геморрагических нарушений не имели.
Генетик заявил, что семейный анамнез напоминает Х-сцепленную аномалию свертывания крови, например гемофилию А или В, а улучшение у брата особенно похоже на гемофилию В с нарушением фактора IX (Лейдена). Для того чтобы подтвердить диагноз гемофилии, необходимо сначала обследовать брата, поскольку диагностика изолированного носительства затруднительна. Пациентка переговорила с братом, и он согласился на обследование.
Изучение его медицинских документов показало, что ему действительно в детстве была диагностирована недостаточность IX фактора, но теперь имел почти нормальные показатели фактора IX в плазме. ДНК-анализ мутации подтвердил, что у него мутация в промоторе гена F9, соответствующая фактору IX Лейдена. Обследование пациентки показало, что она не является носительницей мутации, выявленной у ее брата.
Гемофилия
Заболевание из группы наследственных коагулопатий, обусловленное дефицитом факторов свертывания плазмы крови и характеризующееся повышенной склонностью к геморрагиям – кровотечениям и кровоизлияниям, связанным с нарушением целостности или проницаемости стенок сосудов.
Причины возникновения. Гены, обусловливающие развитие гемофилии, сцеплены с половой Х-хромосомой, поэтому заболевание наследуется по рецессивному признаку по женской линии. Наследственной гемофилией болеют практически исключительно лица мужского пола. Женщины являются проводниками (кондукторами, носителями) гена гемофилии, передающими заболевание части своих сыновей.
Врожденная гемофилия встречается почти у 70 % пациентов. В этом случае наследуется форма и тяжесть гемофилии. Около 30% наблюдений приходится на спорадические, спонтанные формы гемофилии, которые впоследствии становятся наследственными.
Риск передачи заболевания. У здорового мужчины и женщины-кондуктора с одинаковой вероятностью могут родиться как больные, так и здоровые сыновья. От брака мужчины, больного гемофилией, со здоровой женщиной рождаются здоровые сыновья или дочери-кондукторы. Описаны единичные случаи гемофилии у девочек, рожденных от матери-носителя и больного гемофилией отца.
Типы. В зависимости от дефицита того или иного фактора свертываемости крови, различают гемофилию А (классическую), В (болезнь Кристмаса), С и др.
Классическая гемофилия составляет подавляющее большинство (около 85%) случаев синдрома и связана с дефицитом VIII фактора свертывания (антигемофильного глобулина), приводящим к нарушению образования активной тромбокиназы.
При гемофилии В, составляющей 13% случаев заболевания, имеет место недостаток IX фактора (плазменного компонента тромбопластина, фактора Кристмаса), также участвующего в образовании активной тромбокиназы в I фазе свертывания крови.
Гемофилия С встречается с частотой 1-2% и обусловлена недостаточностью XI фактора свертывания крови (предшественника тромбопластина). На остальные разновидности гемофилии приходится менее 0,5% случаев; при этом может отмечаться дефицит различных плазменных факторов.
Симптомы. У новорожденных детей – длительное кровотечение из культи пуповины, подкожные гематомы, кефалогематомы.
У детей первого года жизни – длительные кровотечения в ходе прорезывания зубов, при оперативных вмешательствах. Острые края молочных зубов могут стать причиной прикусывания языка, губ, щек и кровотечений из слизистых оболочек полости рта. Однако в грудном возрасте гемофилия дебютирует редко в связи с тем, что материнском молоке содержится достаточное количество активной тромбокиназы.
Вероятность посттравматических кровотечений значительно возрастает, когда ребенок с гемофилией начинает вставать и ходить. Для детей после года характерны носовые кровотечения, подкожные и межмышечные гематомы, кровоизлияния в крупные суставы. Обострения геморрагического диатеза случаются после перенесенных инфекций (ОРВИ, ветрянки, краснухи, кори, гриппа и др.) вследствие нарушения проницаемости сосудов. Ввиду постоянных и длительных кровотечений у детей с гемофилией имеется анемия различной степени выраженности.
По степени убывания частоты кровоизлияния при гемофилии распределяются следующим образом:
гемартрозы (70—80%) – кровоизлияния в суставы;
гематомы (10-20%) – ограниченнное скопление крови при повреждении органов и тканей;
гематурия (14-20%) – наличие крови в моче;
желудочно-кишечные кровотечения (8%),
кровоизлияния в ЦНС (5%).
Лечение. В лечении гемофилии выделяют два направления – профилактические и «по требованию», в период проявлений геморрагического синдрома.
Профилактическое введение концентратов факторов свертывания крови показано пациентам с тяжелой формой гемофилии и проводится 2-3 раза в неделю для предупреждения развития гемофилической артропатии и прочих кровотечений. При развитии геморрагического синдрома требуются повторные трансфузии препарата. Дополнительно используются свежезамороженная плазма, эритромасса, гемостатики. Все инвазивные вмешательства у больных с гемофилией (наложение швов, удаление зубов, любые операции) проводятся под прикрытием гемостатической терапии.
При незначительных наружных кровотечениях (порезах, кровотечениях из полости носа и рта) используется гемостатическая губка, на рану накладывается давящая повязка, раны обрабатываются тромбином. При неосложненном кровоизлиянии ребенку необходим полный покой, холод, иммобилизация больного сустава гипсовой лонгетой, в дальнейшем – УВЧ, электрофорез, ЛФК, легкий массаж. Больным с гемофилией рекомендуется диета, обогащенная витаминами А, В, С, D, солями кальция и фосфора.
Профилактика. Проведение медико-генетического консультирования супружеских пар, имеющих отягощенный семейный анамнез по гемофилии. Дети, больные гемофилией, всегда должны иметь при себе специальный паспорт, где указан тип заболевания, группа крови и Rh-принадлежность.
Наблюдение. Пациентам показан охранительный режим, профилактика травм; диспансерное наблюдение педиатра, гематолога, детского стоматолога, детского ортопеда и др. специалистов; наблюдение в условиях специализированного гемофильного центра.